Research





DNA arrays and genome sequencing are among the most important technologies in life sciences. In 1989 I published the first paper on computational analysis of DNA arrays (Pevzner, JBSD 1989). Surprisingly, my 30-year old work describing the de Bruijn assembly approach to analyzing DNA arrays is now at the heart of genome assembly algorithms (Pevzner et al., PNAS 2001).

The de Bruijn assembly was first met with skepticism and was viewed as an algorithmic novelty rather than a practical alternative to the classical overlap-layout-consensus approach. Nevertheless, we demonstrated how to apply the de Bruijn approach to next generation sequencing (Chaisson et al., Genome Research 2008, Genome Research 2009) and introduced the notion of the A-Bruijn graphs that generalize the classical de Bruijn graphs (Pevzner et al., Genome Research 2004). As a result, the de Bruijn assembly now dominates the next generation sequencing.

Entire species of bacteria have traditionally been off-limits when it comes to DNA sequencing, because they cannot be cultured. This so-called “dark matter of life" includes the lion's share of bacterial species living on the planet, including microorganisms that could yield new antibiotics. In 2011, my lab developed the first algorithms for single cell sequencing (Chitsaz et al., Nature Biotechnology 2011, Bankevich et al., J. Comp. Biology 2012) that allowed researchers to assemble genomes from DNA extracted from a single bacterial cell.

In 2012 I led the team of researchers from Russia and US to develop SPAdes (Bankevich et al, J. Comp. Biology 2012), the world’s most popular genome assembler with over 20,000 citations to date. We have further turned SPAdes into a Swiss knife of genome assembly by developing plasmidSPAdes (Antipov et al., Bioinformatics 2016), metaSPAdes (Nurk et al., Genome Research, 2017), metaplasmidSPAdes (Antipov et al., Genome Research 2019), metaviralSPAdes (Antipov et al., Bioinformatics 2020), and other assembly tools. Recently, we developed Flye and metaFlye assemblers for long error-prone reads (Lin et al., PNAS 2016, Kolmogorov et al., Nature Biotech, 2019, Kolmogorov et al., Nature Methods, 2019, Bickhart et al., Nature Biotech 2022) as well as LJA assembler for long and accurate reads (Bankevich et al Nature Biotech, 2022).

I was privileged to work with talented students and postdocs on various genome sequencing projects: Dmitry Antipov (postdoc at NIH), Anton Bankevich (professor at Pennsylvania State University), Dima Brinza (senior Director at Ultima Genomics), Christine Boucher (professor at Florida State University), Andrey Bzikadze (bioinformatics scientist a Amazon), Mark Chaisson (professor at USC), Hamid Chitsaz (professor at Colorado State University), Tatiana Dvorkina (bioinformatician at Oxford Nanopores), Earl Hubbel (principal bioinformatician at Grail), Steffen Heber (professor at North Carolina State University), Mikhail Kolmogorov (now inverstigator at NIH) Yu Lin (professor at Australian National University), Paul Medvedev (professor at Pennsylvania State University), Sergey Nurk (bioinformatician at Oxford Nanopores), Son Pham (Chief Scientific Officer at BioTuring), Yana Safonova (professor at Pennsylvania State University), Sing-Hoi Sze (professor at Texas A&M), Haixu Tang (professor at University of Indiana), Glenn Tesler (professor at UCSD), Jeffrey Yuan (scientist at Illumina), and Degui Zhi (professor at University of Texas, Houston).

My laboratory laid foundations of de novo peptide sequencing (Dancik et al., J. Comp. Biology 1999) and applied de novo peptide sequencing to protein identification (Tanner et al., Anal. Chem, 2005, Kim et al., J. Proteome Res., 2008, Kim et al., Molecular and Cellular Proteomics, 2009, 2010, Frank et al., Nature Methods, 2011, Bonissone et al., Molecular and Cellular Proteomics 2012, Jeong et al., Bioinformatics 2013, Kim and Pevzner, Nature Comm., 2014).

We came up with a new approach for assembling mass spectra into proteins using spectral networks (Bandeira et al., Molecular Cellular Proteomics 2007, PNAS 2007) and applied it to develop the first antibody sequencing algorithm (Bandeira et al., Nature Biotechnology, 2008). I co-founded a company Abterra Biosciences, a leader in antibody sequencing and immunoproteogenomics.
My group has advanced to top-down applications of mass spectrometry to intact proteins that enable a new paradigm for protein identification (Liu et al., Molecular and Cell. Proteomics, 2012, J. Proteome Res. 2013, Ansong et al., PNAS 2013, Liu et al., J. of Proteome Res. 2014, Kolmogorov et al., J. of Proteome Res. 2016). Recently, we proposed the first algorithm for the nanopore-based protein identification (Kolmogorov et al., PLoS Computational Biology 2017).
Immunogenomics studies generate millions of reads that sample antibody repertoires and provide insights into monitoring immune response to disease. However, nearly all immunogenomics studies rely on the population-level germline genes rather than germline genes in a specific individual. Personalized immunogenomics (i.e., identifying individual germline genes) is important since variations in germline genes have been linked to various diseases. My lab recently ventured into personalized immunogenomics (Safonova et al., Bioinformatics 2015, Woo et al., J. of Proteome Res. 2015, Cha et al., Mol. Cell Proteomics 2017, Shlemov et al., J. Immunology 2017, Safonova and Pevzner, Frontiers in Immunology 2019, Safonova et al., Genome Research 2020, 2022, Sirupurapu et al., Genome Res. 2022).

I was privileged to work with talented students and postdocs on various proteomics and immunogenomics projects: Nuno Bandeira (now Professor at UCSD), Stefano Bonissone (now Chief Scientific Officer at Digital Proteomics), Vlado Dancik (now scientist at Broad Institute), Nitin Gupta (now professor at Indian Institute of Technology), Kyowon Jeong (now postdoc at University of Tubingen), Sangtae Kim (now CTO at Bertis Biosciences), Xiowen Liu (now professor at Tulane University), Yana Safonova (now professor at Pennsylvania State University), and Dekel Tsur (now professor at Ben-Gurion University).

My laboratory developed the first algorithm for antibiotics sequencing (Ng et al., Nature Methods, 2009) based on the notion of spectral networks (also known as molecular networks). The spectral network approach (Bandeira et al., PNAS 2007) enabled the Global Natural Product Social (GNPS) molecular network that now includes 1000s laboratories working on antibiotics discovery (Wang et al., Nature Biotechnology 2016, Mohimani et al., Nature Chemical Biology, 2017).
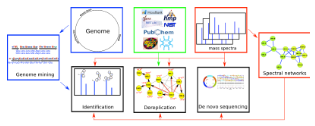
We applied spectral networks and spectral alignments (Pevzner et al., Genome Research 2001, Tsur et al., Nature Biotechnology 2005) to sequencing peptidic antibiotics written in the alphabet of 100s non-standard amino acids that often form cyclic peptides. We developed algorithms for analyzing peptidic antibiotics (Mohimani et al., J. Proteome Res. 2011, ACS Chemical Biology 2014, J. of Natural Products. 2014, Mohimani et al., Nature Chemical Biology, 2017, Gurevich et al., Nature Microbiology 2018, Mohimani et al., Nature Communications 2018, Behsaz et al., Cell Systems 2019, Behsaz et al., Nature Communications 2020) and applied them in various studies aimed at antibiotics discovery (Liu et al., PNAS, 2010, Leao et al., PNAS, 2010, Garg et al., mSystems 2016).

I was privileged to work with talented students and who joined me in various antibiotics projects: Nuno Bandeira (Professor at UCSD), Bahar Behsaz (research scientist at Carnegie Mellon), Alexey Gurevich (professor at Saarland University, Germany), Hosein Mohimani (professor at Carnegie Mellon), and Julio Ng (senior software developer at Microsoft).

If a genome is compared to a continental land-form, then rearrangements represent evolutionary earthquakes that dramatically change the landscape. In 1971, Susumu Ohno introduced the classical Random Breakage Model (RBM) of genome rearrangements that implies that there are no rearrangement hotspots in the human genome. Pevzner and Tesler (PNAS, 2003) refuted RBM and proposed an alternative Fragile Breakage Model (FBM) of chromosome evolution. Shortly afterwards, FBM was reinforced in a joint study with leading biologists (Murphy et al., Science 2005) and expanded into Turnover FBM (Alekseyev and Pevzner, Genome Biology, 2010)

The rebuttal of RBM is based on the notion of the breakpoint graph (Bafna and Pevzner, FOCS 1994) that is now the most popular algorithmic technique for rearrangement studies. The breakpoint graphs analysis resulted in the polynomial algorithms for genome rearrangements (Hannenhalli and Pevzner, FOCS 1995, STOC 1995, J. of ACM 1999) and the first genome rearrangements web servers (Bourque and Pevzner, Genome Research 2002, Alekseyev and Pevzner, Genome Research, 2009).

We contributed genome rearrangement analysis to mouse, rat, and chicken genome sequencing projects (Waterston et al., Nature 2002, Gibbs et al., Nature 2004, Hillier et al., Nature 2004) and further used it to discover the phenomenon of microinversions (Pevzner and Tesler, Genome Research 2003, Chaisson et al., PNAS, 2006). My group was also involved in studies of relationships between rearrangements and duplications (Jiang et al., Nature Genetics 2007, Alekseyev and Pevzner, SODA 2007, SIAM Journal of Computing 2007, Pu et al., Genome Research 2018) and further moved to studies of rearrangements in cancer genomes (Volik et al., Genome Research 2006). We also developed a spliced alignment algorithm (Gelfand et al., PNAS 1996), one of the first comparative genomics approaches in bioinformatics that influenced many follow-up gene prediction algorithms. Recently we developed UniAligner for aligning highly-repetitive genomic regions, such as centromeres (Bzikadze and Pevzner, Nature Methods 2023). My lab has further contributed to analyzing the newly sequenced centromeres in the first telomere-to-telomere sequencing of the human genome (Nurk et al., Science 2022, Bzikadze et al., Genome Res. 2022, Kunyavskaya et al., Genome Res. 2022).

I was privileged to work with talented students and postdocs on various genome rearrangement and comprative genomics projects: Max Alekseyev (professor at George Washington), Vineet Bafna (professor at UCSD), Guillaume Bourque (professor at McGill University), Sridhar Hannenhalli (Principal Scientist at National Cancer Institute), Uri Keich (professor at University of Sydney), Alla Mikheenko (postdoc at Imperial College, London), Qian Peng (Professor at Scripps Research Institute), Son Pham (Chief Scientists at BioTuring), Alkes Price (professor at Harvard), Lianrong Pu (professor at Shandong University), Ben Raphael (professor at Princeton), Sing-Hoi Sze (professor at Texas A&M), and Glenn Tesler (professor at UCSD).
